

 扫码安装网站APP(Android版)
扫码安装网站APP(Android版)
王静雯 李乐
江苏省扬州大学附属中学
摘要:以分析未知序列的反向PCR高考试题为切入点,对测序技术的扩增依赖性、以限制酶识别序列进行引物设计的缺陷、酶切后如何进行环化、环状模板线性扩增机制、测序后的片段分析检验方法等教学疑点进行解读,并拓展了反向PCR的相关应用。
2025 年高考浙江卷的24题以反向PCR(聚合酶链式反应)分析未知序列为情境,综合考查限制酶、DNA连接酶、反向PCR原理及引物设计逻辑,强调对基因工程工具和分子生物学技术的理解与应用。
原试题节选:1,3-磷酸甘油脱氢酶(GPD)是酵母细胞中甘油合成的关键酶。以某假丝酵母菌株为材料,克隆具有高效催化效率的3-磷酸甘油脱氢酶的基因(GPD)。采用的方案是:先通过第1次PCR扩增出该基因的中间部分,再通过第2次PCR分别扩增出该基因的两侧,经拼接获得完整基因的序列,如图1所示,回答下列问题。
图1基因序列扩增方案
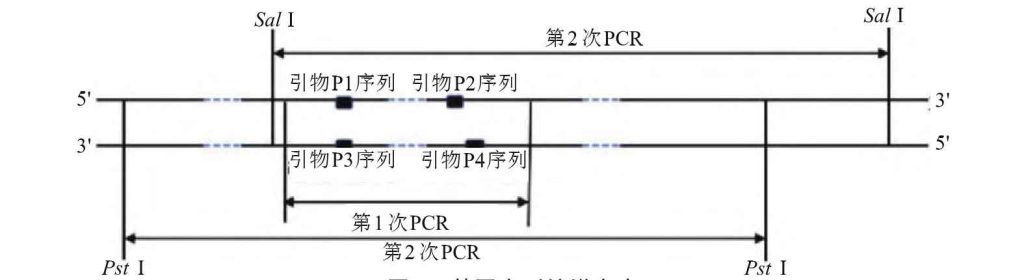
1)分析不同物种的GPD蛋白序列,确定蛋白质上相同的氨基酸区段,依据这些氨基酸所对应的___,确定DNA序列,进而设计1对引物。以该菌株基因组DNA为模板进行第1次PCR,利用凝胶电泳分离并纯化DNA片段,进一步测定PCR产物的序列。在制备PCR反应体系时,每次用微量移液器吸取不同试剂前,需要确认或调整刻度和量程,还需要___。
2)为获得完整的GPD基因,分别用限制性核酸内切酶PstⅠ和SalⅠ单酶切基因组DNA后,各自用DNA连接酶连接形成环形DNA,再用苯酚-氯仿抽提除去杂质,最后加入___沉淀环形DNA。根据第1次PCR产物测定获得的序列,重新设计1对引物,以环形DNA为模板进行第2次PCR,最后进行测序。用于第2次PCR的1对引物,其序列应是DNA链上的___(A.P1和P2B.P3和P4C.P1和P4D.P2和P3)。根据测序结果拼接获得完整的GPD基因序列,其中的启动子和终止子具有___功能。
该试题以创新性情境设计为依托,聚焦生物学核心素养的深度考查。本题表述高度凝练,蕴含多维信息架构。其中,反向PCR技术原理的呈现尤其值得关注,因其涉及环状DNA扩增机制和引物方向性设计等抽象概念,暴露出了中学教学实践中的一些认知盲区:部分教师对关键技术要点(如限制性内切酶的作用时序、DNA连接酶的热稳定性)的理解存在偏差,这种认知偏差直接导致试题命制与解析过程中科学严谨性的缺失。
反向PCR作为PCR技术的变体,契合高中生物学课程标准的要求,能完善学生对PCR技术体系的认知;深化学生对核酸扩增原理的理解,培养学生的辩证思维能力,其在实验设计、结果分析等环节具有不可替代的教学价值。浙江高考题再次考查反向PCR相关知识点,进一步印证了其在高考中的重要地位。基于此,本研究通过文献分析与技术原理溯源,系统解析反向PCR的关键技术环节,并对相关问题进行解读。
1 已经获得基因片段不直接进行测序的原因
传统Sanger测序需满足μg至ng级模板量或pmol/L级引物浓度,且依赖已知侧翼序列设计引物。本试题中,假丝酵母经限制酶(如PstⅠ或SalⅠ)单酶切整个基因组DNA后,会产生很多个长度不一的DNA片段,可以分为包含完整目的基因和不含目的基因的2类片段。因未纯化导致非目的片段混杂,模板浓度不足且侧翼序列未知,无法满足Sanger测序条件。虽然Oxford Nanopore等单分子测序技术无须预知引物位点且模板需求低,但其单碱基原始错误率(约5%)、单位数据成本(Illumina测序的3~5倍)及读长数据处理对专用工具(如Medaka、Canu)的依赖性限制了其广泛应用。当前主流平台中,Illumina等2代技术须通过PCR扩增(如桥式扩增成簇)富集模板,而3代技术虽支持单分子测序,但低起始量样本仍需要全基因组扩增。如表1构建的测序技术代际特点与扩增依赖性矩阵所示,当前主流测序平台仍依赖PCR扩增实现模板富集,这从根本上决定了PCR在该测序流程中的重要性。
表1测序技术代际特点与扩增依赖对比

*注:第4代技术侧重空间原位检测,读长非主要指标
2 不能直接根据基因两侧的限制酶序列设计引物进行扩增的原因
试题呈现的图中可以看出GPD基因两端分别存在PstⅠ和SalⅠ的酶切位点,能否直接以酶切位点序列设计引物进行PCR?在进行PCR时,引物设计的相关原则及注意事项较多,直接使用限制酶识别序列设计引物不满足相关要求。
2.1 引物设计规范冲突
引物有效退火需要满足18~30 bp的长度要求且Tm≥55℃,但限制酶识别序列仅6 bp,其Tm值约12℃,结合特异性指数不足标准值的15%。且短引物在复杂基因组中随机匹配概率极高,这会使引物与模板发生大量非特异性结合,进而引发诸多非特异性扩增。
2.2 二级结构对扩增的阻碍性
GPD基因两侧存在未知序列,且限制酶位点周边序列常富含重复元件使模板形成稳定二级结构(如发夹或茎环),导致覆盖引物结合位点,使引物无法接触模板。即使部分二级结构在高温变性步骤中被打开,在降温退火时可能迅速重新形成,与引物退火形成竞争。即使引物与模板勉强结合,二级结构也可能阻碍聚合酶的移动,导致扩增提前终止。因此,在实际操作时,常避开此类区域。
3 酶切后如何进行环化
环化过程要考虑2个问题:1)限制酶还在反应体系中,此时加DNA连接酶无法将酶切后的GPD基因环化。2)因为分别对GPD基因进行单酶切后其两端的黏性末端相同,故酶切后的2类片段通过DNA连接酶进行连接时,不仅会发生分子内环化(单个片段首尾相连形成环状),也会发生分子间连接(多个片段首尾相连形成线性或环状多聚体)。在这些连接产物中,只有一小部分是目标环状分子,即包含了目标基因(GPD)及其上游(PstⅠ切点)或下游(SalⅠ切点)未知侧翼序列的环状DNA。如何减少非特异性环状DNA分子出现的概率?高中阶段的题目暂未考查相关知识,导致许多教师教学时忽视了这2个问题,但这是科研操作中必须要考虑到的问题。在高中生物学教学中讲解这些问题,能使学生理论联系实际,深入理解分子生物学知识,培养学生解决问题的能力和科学思维,为未来实验操作和科研工作奠定基础。因此,通过查阅资料,了解到实际实验过程规避以上问题的方法。
针对第1个问题,在实际操作时会在限制酶酶切之后进行65℃左右灭活,时长15~20 min,确保限制酶失活后再加入DNA连接酶进行连接。或通过有机溶剂(苯酚/氯仿)抽提去除蛋白质类酶,纯化DNA后重新调整缓冲体系进行连接反应。在该试题3)问的描述中则是用DNA连接酶连接形成环形DNA后,再用苯酚-氯仿抽提除去杂质。部分教师误认为该操作是为了区分不同类型的环状分子,但实则是纯化所有连接后形成的环状DNA混合物(去除蛋白质、盐分等杂质)。真正的筛选发生在第2次PCR:通过设计特异性引物,这些引物只能与目标环状分子上的已知序列结合(且方向正确),从而选择性地扩增出包含相邻未知序列的线性片段。非目标环状分子(如其他基因组片段环化形成的环、串联体形成的环)由于缺乏引物结合位点或引物方向不匹配,无法被有效扩增,或者在后续的电泳/测序分析中被排除。
针对第2个问题,在实际操作时会降低DNA质量浓度(如低于10 ng/μL),促进分子内环化(单个片段自身连接),抑制分子间连接(多个相同片段串联),或对连接时间进行控制,如缩短连接时间(如4 h),减少多聚体形成概率。
4 扩增得到何种目标产物
由试题1)可知,第1次扩增后测序分析得到了GPD基因中间的一段序列,将其定为已知序列。再根据第3节可知,后续的步骤依次为:使限制酶失活、加DNA连接酶进行连接反应、用苯酚-氯仿抽提除去杂质、根据中间已知序列设计特异性引物进行扩增。第2次扩增是为了获得已知序列两侧的未知序列,所以第2次扩增应该为反向扩增,对应的引物应该设计为序列2、3的反向引物,即3′端朝向外侧。因DNA聚合酶只能将游离的脱氧核苷酸连接到已有核酸片段的3′末端,所以在其作用下游离脱氧核苷酸会不断连接到特异性引物的3′端进行反向延伸。又因DNA连接酶在上一步的操作中被大量去除,少量残留的DNA连接酶在PCR的高温条件下失活,所以第2轮PCR的第1个循环延伸到引物的5′端后,无法催化最后一个磷酸二酯键的形成而出现缺口,结果形成2个带有断口的开环DNA分子。后续再以该开环DNA分子为模板经过第2、3个循环便能得到缺失部分已知序列的线性DNA分子。基于以上推理,能分析出添加序列为P2、P3的引物后所扩增出的产物,图2以PstⅠ酶切环化后的扩增为例进行演示。
由图2可知,一个环状DNA第3次扩增可以得到2条目的片段,所以扩增结束会得到大量的目的片段,且满足公式2n-2n(n代表循环次数)。由此,可推断若选择P1、P4序列的引物,在Taq酶作用下只能进行正向扩增,则最终会得到大量引物及其内侧的已知序列,如图3所示。
图2 PstⅠ酶切环化后选择序列为P2、P3的引物(用引物3、4代替)的扩增示意图
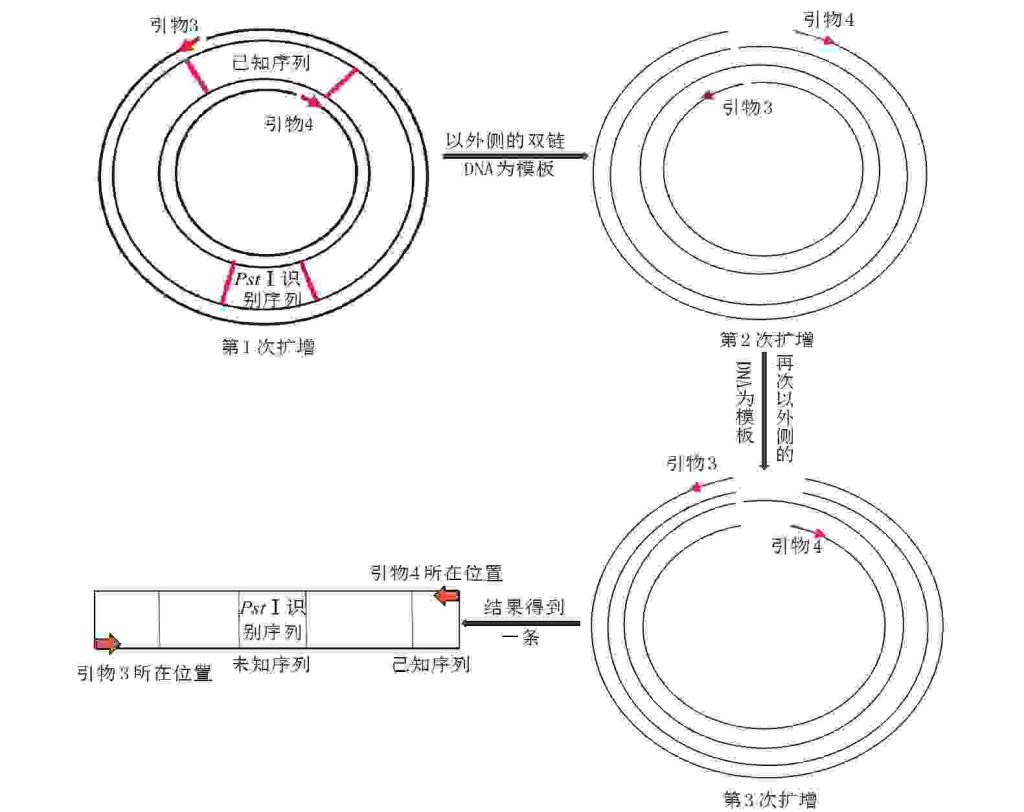
图3 选择序列为P1、P4的引物(用引物1、2表示)扩增结果示意图
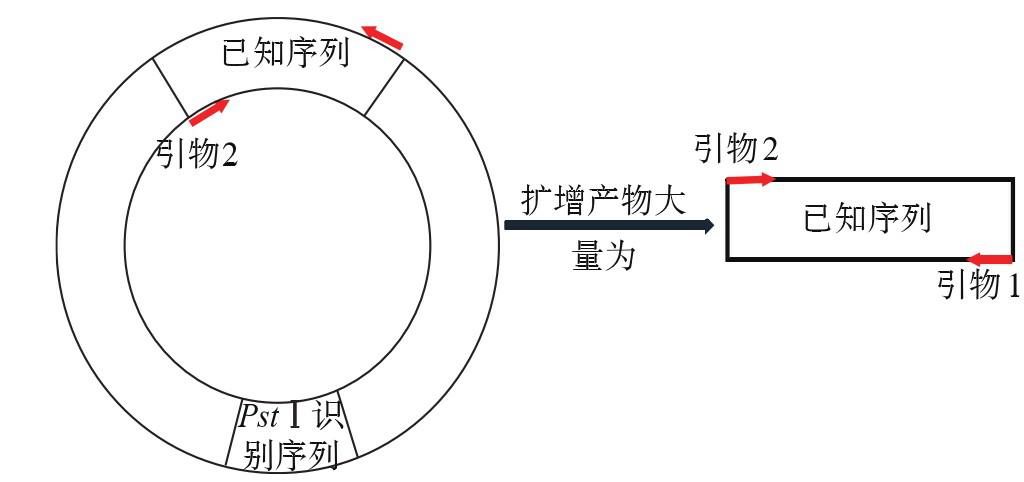
5 如何对GPD基因片段进行测序、分析并比对
通过前面的扩增结果可知,第2次PCR的扩增产物主要为缺失部分已知序列的2种片段。可分别通过电泳的方法将其筛选出来。此时可以用3代测序技术(如Oxford Nanopore)直接对原始DNA模板进行单分子测序,无须扩增;或用2代测序(如Illumina)先通过PCR扩增(桥式扩增)生成DNA簇,在扩增过程中进行边合成边测序。用1代测序(Sanger法)则须通过PCR扩增目标片段后,再对纯化产物进行测序,所用的引物可以与前面选择的反向引物相同。测序结束得到了扩增产物的完整序列,须对测序片段进行分析,过程如图4所示。
图4测序结果的分析示意图
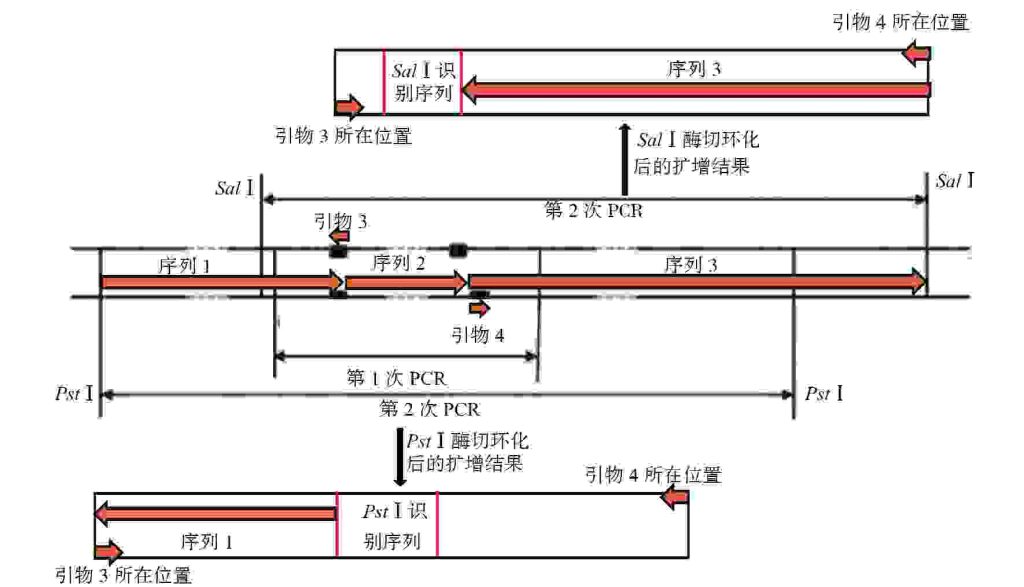
由图4可知,第1次PCR扩增是为得到该基因的中间部分,经测序分析后得到其序列,再根据其序列选择合适位置设计反向引物进行第2次PCR,从而分别扩增出该基因的两侧,再测序得到该基因两侧的序列,进行拼接后即获得完整基因的序列。具体的拼接逻辑如下:如图4所示,GPD基因从左到右分为3个序列,序列1为左侧PstⅠ识别序列到反向引物3的5′端对应的位置;序列2为反向引物3和4对应位置之间的序列;序列3为反向引物4的5′端对应的位置到最右侧的SalⅠ识别序列,序列1、3均不包含限制酶识别序列。引物3的5′端在图示扩增产物的最左侧,而PstⅠ识别序列在扩增产物的内部,由于限制酶识别序列是已知的,故在对测序后的序列分析时可知,由内部PstⅠ识别序列指向扩增产物的最左侧即为GPD基因的序列1,同理分析序列3应该为图示SalⅠ酶切环化后的扩增产物的最右侧,指向内部SalⅠ识别序列。综上所述,第1次PCR后测序得到了序列2,第2次PCR后经测序便能得到序列1和序列3,将其按照顺序拼接便能得到完整的GPD基因。获得完整的GPD基因序列后,利用生物信息学工具,特别是BLAST(basic local alignment search tool),将其与公共数据库中的基因组序列进行比对,确认该序列是否为目标基因,并确认其在染色体上的具体位置。以常见的白假丝酵母菌GPD1基因为例进行BLAST分析演示:假设通过测序后分析获得了一段序列,将其提交至BLAST服务器,选择blastn程序(用于核酸序列比对)和合适的数据库,其他参数设置默认,最后点击BLAST,结果如图5所示。
图5 blastn比对关键结果截图(英译中版)
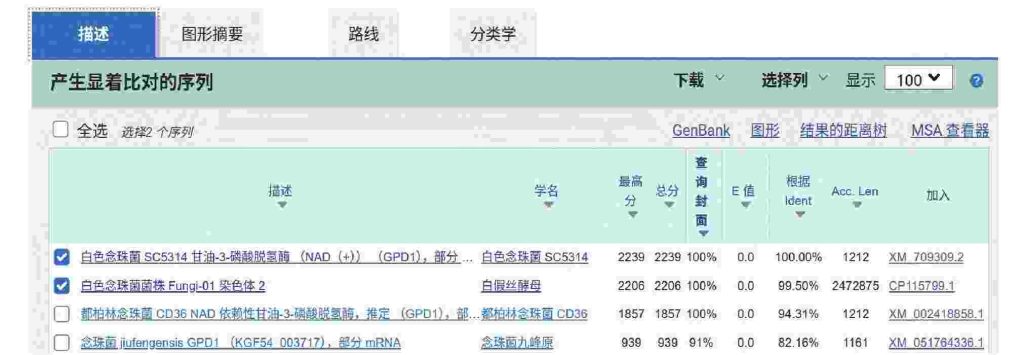
图5展示了针对获得的白假丝酵母菌基因序列进行的blastn比对关键结果。分析首先确认了目标基因:序列与白色念珠菌(白假丝酵母)标准菌株SC5314的甘油-3-磷酸脱氢酶基因(GPD1)的mRNA序列完美匹配(查询覆盖率100%,一致性为100%,E值为0.0),证明所得序列即为目标基因GPD1。更重要的是,该序列与白假丝酵母菌株Fungi-01的完整染色体2序列也近乎完美匹配(查询覆盖率为100%,一致性为99.5%,E值为0.0)。这一关键匹配将GPD1基因精确定位于该菌株的染色体2上,为理解其基因组环境(如侧翼调控区域、邻近基因)奠定了基础。该结果清晰展示了反向PCR测序技术结合生物信息学分析(BLAST)在克隆基因身份验证及染色体精确定位中的强大作用。
6 反向PCR的应用
在高中生物学教学中,反向PCR技术的学习能够深化学生对分子生物学技术的理解,但只学习技术不了解实际应用会使学生陷入技术原理的机械记忆,难以建立“技术发明-科学问题-现实需求”的认知闭环,导致知识碎片化、迁移能力缺失,无法应对真实科研情境中的相关问题。因此,在原理学完之后,需要及时补充相关应用。利用无缝克隆技术构建重组质粒时,通常利用反向PCR将环状DNA变成线状DNA,暴露同源序列便于目的基因与载体进行同源重组;在克隆基因启动子时(如猪H-FABP基因5′调控序列),反向PCR通过环化酶切后的基因组DNA,利用已知序列设计反向引物,成功扩增出上游未知区域;在疾病研究中(如乳腺癌细胞9p21缺失断点定位),该技术通过连接断裂点两侧序列形成环状模板,精准解析染色体结构变异。此外,反向PCR还被用于构建蛋白截短体质粒(如TRAF6功能域研究)和分离组织特异性启动子(如小麦花粉特异基因PSG076)。这些应用案例覆盖了农业育种、医学诊断和基础研究等多个领域,体现了技术的前沿性和实用性。
来源:王静雯,李乐.基于高考试题解读和拓展“反向PCR测序技术”[J].生物学通报,2025,60(12):67-71.



近期评论